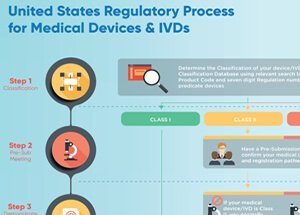
Los productos sanitarios pueden definirse como de clase I, clase II o clase III, en función del nivel de riesgo que representan. Los únicos dispositivos que requieren la aprobación de la FDA son los dispositivos de Clase III, la categoría de mayor riesgo. Para que un dispositivo de Clase III sea aprobado por la FDA, el fabricante debe proporcionar evidencia de su seguridad y efectividad.
La aprobación de la FDA no es necesaria para los dispositivos de Clase I y Clase II. Todos los fabricantes de dispositivos que no están exentos, en virtud de la Ley FD&C, deben presentar una notificación previa a la comercialización (510(k)) a la FDA en su lugar. El propósito de esta notificación es proporcionar evidencia a la FDA de que este dispositivo es al menos tan seguro y efectivo como un dispositivo que ya está en el mercado. Si la FDA determina que el dispositivo es sustancialmente equivalente a otros dispositivos que ya están en el mercado, puede ser comercializado en los Estados Unidos. No se trata de la misma homologación exigida para los dispositivos de Clase III.
Los fabricantes de dispositivos deben registrarse en la FDA y hacer un listado de sus dispositivos . Sin embargo, ni el registro ni el listado del dispositivo indica la aprobación de la FDA del fabricante o del producto.