Skip to content
Skip to content High risk (Class III) devices require premarket approval by the FDA before they can be sold in the U.S. This can be a complicated process, as it requires the submission of clinical evidence of the safety and effectiveness of the device. Cosmereg offers a full premarket approval application service, ensuring that your application meets all of the FDA requirements.
FDA Medical Devices Regulations 21 cfr 803
The term ‘Medical Device’ refers to an enormous range of products, from protective clothing for healthcare professionals to software, to surgical tools and more. In the U.S. Medical Devices are regulated by the Food and Drugs Administration (FDA). Certain medical devices required FDA approval for their sale. Below the FDA medical devices regulations 21 cfr 803 processes.
- Accurate Review
- Fast Submission
- 10 years experience
U.S. FDA MEDICAL DEVICES REGULATIONS
MEDICAL DEVICE ESTABLISHMENT REGISTRATION AND DEVICE LISTING
Manufacturers of Medical Devices of any class must register with the FDA. Foreign manufacturers must also appoint a U.S. Agent to act as an intermediary between the manufacturer and the FDA. Cosmereg can support your business in registering establishments. We also offer a full U.S. Agent service for our foreign clients.
PREMARKET NOTIFICATION 510 (K)
Many medical devices require a letter of substantial equivalence from the FDA, stating that the device intended for sale is as safe and effective as other similar devices on the market. This is obtained by applying for pre-market approval (501k). Cosmereg can support your business in the application process following the FDA medical devices regulations and21 cfr 803.

PMA (PREMARKET APPROVAL)

LABELING REVIEW
In order to meet the requirements for premarket approval, clinical trials of medical devices must be undertaken. A special permission exists for the use and sale of devices in this context, known as ‘Investigational Device Exemption’ (IDE). Cosmereg can support your business in applying for this exemption and 21 cfr 803.
INVESTIGATIONAL DEVICE EXEMPTION (IDE)
In order to meet the requirements for premarket approval, clinical trials of medical devices must be undertaken. A special permission exists for the use and sale of devices in this context, known as ‘Investigational Device Exemption’ (IDE). Cosmereg can support your business in applying for this exemption and 21 cfr 803.

DE NOVO SUBMISSION
Entirely new devices are automatically categorized as Class III devices, as it is not possible to complete an assessement of equivalence with other devices. If your device represents a low risk, you can demonstrate this to the FDA through a ‘De Novo’ application (513[g]). Cosmereg can support your company thorough this process to ensure that your device is appropriately classified as swiftly as possible.
Need guidance selecting the right service?
Contact us today for a free consultation
FAQ
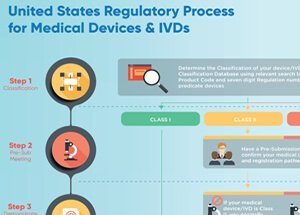
21 cfr 803 Medical devices can be defined as Class I, Class II or Class III, based on the level of risk that they represent. The only devices that require approval by the FDA are Class III devices – the highest risk category. In order for a Class III device to be approved by the FDA, the manufacturer must provide evidence of its safety and effectiveness.
FDA approval is not required for Class I and Class II devices. All manufacturers of devices that are not exempt, under the FD&C Act, must instead submit a premarket notification (510(k)) to FDA instead. The purpose of this notification is to provide evidence to the FDA that this device is at least as safe and effective as a device that is already on the market. If the FDA finds that this device is substantially equivalent to other devices that are already on the market, it can be marketed in the US. This is not the same as the approval that is required for Class III devices.
Device manufacturers must register with FDA and list their devices. However, neither registration nor listing the device indicates FDA approval of the manufacturer or the product.
FDA Medical devices Flow Chart