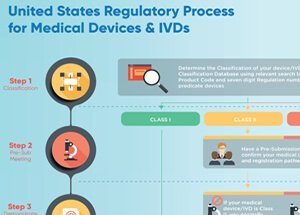
I dispositivi medici possono essere definiti come Classe I, Classe II o Classe III, in base al livello di rischio che rappresentano. Gli unici dispositivi che richiedono l’approvazione della FDA sono dispositivi di Classe III – la categoria di rischio più elevata. Affinché un dispositivo di Classe III sia approvato dalla FDA, il produttore deve fornire prove della sua sicurezza ed efficacia.
L’approvazione della FDA non è richiesta per i dispositivi di Classe I e Classe II. Tutti i produttori di dispositivi che non sono esentati, ai sensi della legge FD & C, devono invece inviare alla notifica di pre-vendita (510 (k)) alla FDA. Lo scopo di questa notifica è di fornire alla FDA evidenza che questo dispositivo è almeno altrettanto sicuro ed efficace di un dispositivo che è già sul mercato. Se la FDA rileva che questo dispositivo è equivalente ad altri dispositivi già presenti sul mercato, può essere commercializzato negli Stati Uniti. Questo non è lo stesso dell’omologazione richiesta per i dispositivi di Classe III.
I produttori di dispositivi devono registrarsi presso la FDA e elencare i propri dispositivi. Tuttavia, né la registrazione né l’elenco del dispositivo indicano l’approvazione FDA del produttore o del prodotto.